Définition
HERNS (Hereditary Endotheliopathy, Retinopathy, Nephropathy and Stroke), HVR (Hereditary Vascular Retinopathy) et CRV (CerebroRetinal Vasculopathy) sont les dénominations d’une même affection autosomique dominante responsable d’une atteinte vasculaire rétinienne et de la substance blanche cérébrale. Cette affection est liée à des mutations du gène TREX1, situé sur le bras court du chromosome 3. Depuis l’identification du gène impliqué, le terme commun de RVCL (autosomal dominant Retinal Vasculopathy with Cerebral Leukodystrophy) a été proposée.
Epidémiologie
L’implication du gène TREX dans la survenue de RVCL est récente. La prévalence est inconnue. Seules quelques familles ont été rapportées à ce jour.
Description clinique et pronostic
Le spectre clinique et l’histoire naturelle de cette affection ne sont pas encore parfaitement connus. Le diagnostic de HERNS doit être évoqué devant la survenue chez un adulte d’âge moyen d’une atteinte rétinienne et/ou cérébrale.
Au cours de cette affection, les symptômes visuels peuvent précéder les déficits neurologiques. Il s’agit le plus souvent d’une baisse de l’acuité visuelle à début central en rapport avec une rétinopathie vasculaire associant occlusions capillaires, télangiectasies et microanévrysmes de topographie maculaire voire juxta-fovéolaire. Parfois les lésions rétiniennes débutent en périphérie et les symptômes visuels sont moins sévères. En l’absence de cause identifiée, ces anomalies justifient la réalisation d’une imagerie cérébrale à la recherche de lésions de la substance blanche parfois silencieuses.
Les lésions cérébrales peuvent être limitées à des hypersignaux de la substance blanche et à des infarctus de petite taille ou prendre dans certains cas une allure pseudotumorale avec prise de contraste en IRM. Ces lésions à l’origine d’un effet de masse sont souvent bruyantes et révélées par un déficit neurologique focal, une hypertension intracranienne, et/ou des crises d’épilepsie. D’autres symptômes neurologiques (migraines sans ou avec aura, dépression, troubles psychiatriques) peuvent être observés. L’évolution est dans la majorité des cas péjorative et aboutit au décès des patients en 5 à 10 ans.
Chez certains patients, l’atteinte rétinienne ou cérébro-rétinienne est associée à une néphropathie glomérulaire avec insuffisance rénale et protéinurie. Des phénomènes de Raynaud ont également été rapportés dans certaines familles.
Etiologie/physiopathologie
TREX1 est impliqué dans le remodelage des extrémités de l’ADN réduisant le nombre de fragments aberrants d’ADN au niveau intracellulaire. Les mutations impliquées dans le HERNS entraînent un décalage du cadre de lecture (« frameshift ») après les sites actifs de la protéine, avec pour conséquence la fabrication d’une protéine TREX1 tronquée. Ceci n’altère pas l’activité de la protéine, mais modifie sa distribution subcellulaire. En microscopie électronique, l’examen ultrastructural révèle un dédoublement de la membrane basale en multiples couches au niveau des capillaires et des artérioles du cerveau, des reins, de la peau et de certaines muqueuses. TREX1 est également impliqué dans d’autres affections très différentes. Des mutations homozygotes mais aussi hétérozygotes de ce gène sont responsables de syndromes d’Aicardi-Goutières associant une encéphalopathie sévère du nourrisson, des calcifications cérébrales et une lymphocytose du LCR. Des mutations de TREX1 ont également été identifiées dans une forme autosomique dominante de lupus cutané avec engelures et chez environ 2% des patients ayant un lupus érythémateux disséminé.
Diagnostic (critères-méthodes)
Le mode de transmission de la maladie est autosomique dominant (atteinte de même fréquence des sujets de sexe masculin et féminin, 50% des enfants issus d’un sujet atteint ayant l’anomalie génétique). Un interrogatoire à la recherche d’antécédents évocateurs de l’affection chez d’autres membres de la famille est indispensable. Une histoire de rétinopathie ou de symptômes neurologiques d’allure centrale chez un apparenté du 1er ou du 2ème degré peut faire évoquer le diagnostic.
Le diagnostic est le plus souvent évoqué devant une atteinte cérébrale ou rétinienne évocatrice.
L’atteinte cérébrale est le mieux visualisée en IRM. L’examen doit au minimum comprendre des séquences pondérées en T1, T2, FLAIR et écho de gradient.
Cet examen permet de rechercher les anomalies décrites au cours du syndrome :
- lésions de la substance blanche cérébrale
- infarctus cérébraux de petite taille
- lésions d’allure pseudo-tumorale prenant le contraste
La rétinopathie vasculaire est diagnostiquée au cours de l’examen du fond d’œil par un ophtalmologiste et montre des occlusions capillaires, des télangiectasies et/ou des microanévrysmes de topographie maculaire. Les photographies de la rétine (à l’aide d’un appareil appelé rétinographe) complété parfois par l’angiographie des vaisseaux de la rétine (injection d’un produit par une veine du bras pour mieux voir les vaisseaux) permettent de mieux analyser ces anomalies.
La réalisation d’un test génétique est indispensable pour poser avec certitude le diagnostic de RVCL. Des mutations de la partie C-terminale de l’exon unique du gène TREX1 (3-5 Repair Exonuclease 1) situé sur le bras court du chromosome 3 sont recherchées.
Le diagnostic génétique est possible avant l’apparition des symptômes de la maladie pour les membres d’une famille atteinte. Cependant, la réalisation d’un test génétique chez un sujet sain n’ayant aucune manifestation clinique de la maladie et n’ayant eu aucun examen préalable n’est effectuée que dans le cadre d’une consultation spécialisée et multidisciplinaire. Après une évaluation neurologique (neurologue), psychologique (entretien avec un ou une psychologue) et une consultation de génétique (généticien), la demande du patient est évaluée de façon collégiale et un délai de réflexion est proposé avant la réalisation d’un prélèvement sanguin. Un suivi clinique et psychologique est toujours proposé après l’annonce des résultats.
Prise en charge
Aucun traitement préventif spécifique n’est connu à ce jour. L’utilisation de corticoïde par voie orale ou parentérale peut s’avérer transitoirement efficace sur l’œdème périlésionnel associé aux lésions pseudo-tumorales. Cependant, ni la corticothérapie, ni la chirurgie d’exérèse ne permettent de stopper le cours de la maladie.
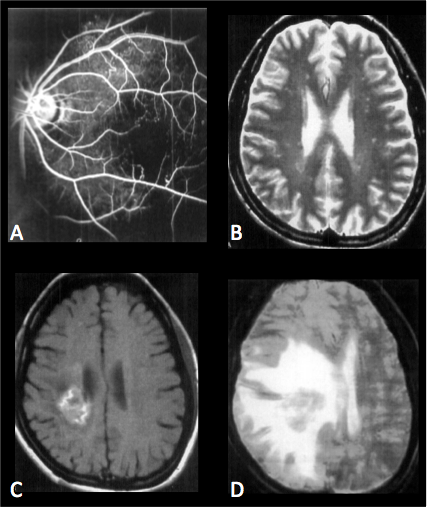
Figure : RVCL: Anomalies des artères rétiniennes à l’angiographie et lésions cérébrales visualisées en IRM.
Légende:
A: Angiographie à la fluorescéine au temps veineux réalisée en raison de symptômes visuels évoluant depuis l’âge de 31 ans et révélant des aires de non perfusion capillaire dans la région maculaire, des vaisseaux dilatés et tortueux associés à des télangiectasies et des shunts capillaires.
B: IRM (FLAIR) réalisée à l’âge de 31 ans en l’absence de symptôme neurologique révélant des hypersignaux punctiformes de la substance blanche.
C: IRM (T1 après injection de gadolinium) réalisée à l’âge de 36 ans en raison d’une maladresse des membres supérieur et inférieur gauche et révélant une lésion d’allure pseudotumorale.
D: IRM (Densité de protons) réalisée un mois plus tard en raison d’un tableau d’hypertension intravranienne confirmant une augmentation de taille de la lésion pseudotumorale et de l’œdème périlésionnel associé à une effet de masse important.
Source: Hereditary endotheliopathy with retinopathy, nephropathy, and stroke (HERNS), J. Jen and al., Neurology 1997;49:1322-1330.