Définition
CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy) est une maladie héréditaire autosomique dominante touchant les petites artères cérébrales, responsable d’infarctus sous-corticaux et d’une atteinte de la substance blanche (leucoencéphalopathie), due à différentes mutations du gène NOTCH 3 localisé sur le chromosome 19.
Epidémiologie
D’abord décrite en Europe, la maladie a été maintenant observée dans des familles d’origine ethnique très différente, sur tous les continents. On dénombre actuellement plus de quatre cents familles en Europe. Il n’existe pas encore de véritable étude épidémiologique consacrée à CADASIL en France. Les auteurs d’une étude réalisée dans l’ouest de l’Ecosse en 2002 ont dénombré 22 patients atteints de CADASIL issus de sept familles au sein d’une population de 1.418.990 habitants. En considérant également les apparentés de ces patients, à risque d’être porteur du gène muté, ils ont estimé la prévalence à 4.15/100.000 habitants. La fréquence de la maladie est cependant encore probablement sous-estimée.
Description clinique
Les premières manifestations cliniques, observées chez 1/3 des patients, sont la survenue de crises de migraine avec aura débutant le plus souvent avant 45 ans et s’améliorant avec l’âge. Les accidents vasculaires (AVC) ischémiques (infarctus cérébraux) sont observés chez 60 % des patients le plus souvent aux alentours de cinquante ans. On note également des troubles cognitifs (troubles de la concentration, de l’attention, troubles de la mémoire), d’importance variable: présents très tôt dans l’évolution de la maladie, ils sont rarement handicapant avant l’âge de 50 ans. Ces troubles cognitifs peuvent être à l’origine d’une altération de la vie sociale et aboutir à une démence à la phase terminale de la maladie. Environ 20% des patients de plus de 55 ans sont concernés. La survenue de troubles de la marche et de l’équilibre est également fréquente et concernent la moitié des patients de plus de 55 ans. On retrouve également, dans 10 à 20% des cas des épisodes de troubles psychiatriques et dans 5 à 10 % des cas une comitialité (des crises épileptiques).
Les crises de migraine avec aura (crises de migraine accompagnées de signes neurologiques) sont rapportés par un patient sur trois. La fréquence des crises est extrêmement variable, de deux épisodes par semaine à un tous les 3 ou 4 ans. Les symptômes de l’aura sont, par ordre de fréquence, visuels, sensitifs, aphasiques ou moteurs. L’aura visuelle se manifeste sous des formes variables : il s’agit le plus fréquemment d’un scotome scintillant, moins souvent d’une vision brouillée ou d’une hémianopsie latérale homonyme. Les troubles de la parole au cours des crises de migraine avec aura se résument souvent à des difficultés d’expression avec une réduction de la fluence verbale.
Plus de la moitié des patients présentent des crises avec des auras atypiques : crises avec aura à début brutal par exemple, crises de migraine « basilaire » ou crises de migraine « hémiplégique ». Dans quelques cas, des crises extrêmement sévères comme celles observées au cours de la migraine hémiplégique familiale, sont observées à l’origine d’épisodes avec confusion, troubles de la vigilance, coma, hyperthermie pouvant durer plusieurs heures, parfois plusieurs jours.
Environ, 60 % des patients rapportent la survenue d’un accident ischémique cérébral transitoire (déficit neurologique régressant en moins de 24h) ou constitué (déficit neurologique permanent) durant l’évolution de la maladie. Il s’agit le plus souvent de manifestations en rapport avec un infarctus de petite taille au niveau du cerveau, à l’origine de manifestations classiques (syndrome lacunaires dues à l’occlusion d’une petite artère: déficit sensitif pur, déficit moteur pur ou déficit sensitivomoteur d’un hémicorps ou hémiparésie ataxie). Ces infarctus cérébraux peuvent survenir en l’absence de tout facteur de risque vasculaire habituel (hypertension artérielle, diabète, hypercholestérolémie).
Les troubles de l’humeur sont observés chez un patient sur cinq. Ils peuvent être inauguraux (jusqu’à 10 % des patients) et conduire à une erreur ou un retard diagnostique. Certains patients présentent des symptômes dépressifs sévères évoquant une mélancolie, qui alternent rarement avec des épisodes maniaques (pouvant faire envisager le diagnostic de trouble bipolaire). L’apathie (perte des motivations) est un signe fréquent de la maladie en rapport avec la localisation des lésions au niveau cérébral. Elle n’est pas toujours secondaire à la dépression.
Les troubles cognitifs (troubles des fonctions exécutives, troubles de l’attention, troubles de la mémoire) sont extrêmement fréquents mais de sévérité très variable au cours de la maladie. L’altération des fonctions exécutives (tâches de planification, d’anticipation, d’ajustement, d’autocorrection et de souplesse mentale) est la plus fréquemment et précocement observée et peut être difficilement perceptible de nombreuses années compte tenu du retentissement le plus souvent minime ou absent dans la vie quotidienne. L’atteinte des fonctions exécutives est fréquemment associée à des troubles de l’attention et de la mémoire de travail. Progressivement, avec l’âge, le déclin peut devenir plus sévère avec l’apparition d’une apathie souvent au premier plan et de déficits des fonctions instrumentales (retentissant sur des tâches telles que le dessin, l'écriture) en faveur d’une atteinte cérébrale diffuse. On ne retrouve cependant que rarement une aphasie sévère (troubles du langage), une apraxie (trouble du comportement gestuel volontaire) ou encore une agnosie (trouble de la reconnaissance des objets, des personnes ou lieux dans trouble visuel) fréquemment observés au cours de la maladie d’Alzheimer. La mémoire sémantique (en rapport avec les connaissances) et la reconnaissance sont souvent préservées. Le déclin cognitif apparait le plus souvent progressivement, le plus souvent en l’absence d’évènements ischémiques, cette évolution peut ainsi évoquer une maladie dégénérative. Parfois, l’aggravation se fait de façon brutale ou par à-coups.
La démence (trouble cognitif retentissant sur la vie du sujet et conduisant à une perte d’autonomie) est observée chez 20% des patients âgés de plus de 55 ans. Elle est fréquemment associée à d’autres signes de gravité de la maladie : troubles de la marche, incontinence urinaire et parfois à un syndrome pseudo-bulbaire (troubles de la déglutition, rire ou pleurer spasmodiques).
L’expression clinique diffère en partie en fonction du sexe. Les femmes sont plus concernées par la survenue de crises de migraine avec aura alors que les hommes ont plus souvent des infarctus cérébraux. Le niveau d’éducation, lorsqu’il est élevé, est associé à une prévalence d’AVC diminuée et à de meilleurs scores cognitifs.
Enfin, malgré l’atteinte diffuse des petites artères de tous les organes, les manifestations de la maladie sont exclusivement cérébrales.
Evolution clinique et pronostic
Le profil évolutif classique de la maladie se caractérise par l’apparition de crises de migraine avec aura au cours de la troisième décennie, par la survenue d’accidents ischémiques cérébraux transitoires ou constitués, une dizaine d’années plus tard, et par l’installation progressive des troubles cognitifs, de troubles de l’équilibre et de la marche vers la soixantaine. La perte d’autonomie, le handicap moteur et cognitif sont fréquents après 70 ans (Figure 1)
Ce profil évolutif est cependant très inconstant en raison de la grande variabilité évolutive de la maladie qui peut être observée entre parfois plusieurs membres d’une même famille (avec donc la même anomalie génétique). Ainsi, dans certains cas, la maladie peut être à l’origine d’un handicap précoce vers l’âge de 40 ans. A l’inverse, les premières manifestations de la maladie peuvent apparaitre après l’âge de 60 ans et rester minimes chez d’autres individus.
Enfin, la survenue d’un accident ischémique cérébral n’est pas un facteur pronostic essentiel à lui seul. La progression de la symptomatologie neurologique est avant tout progressive et s’accélère à partir d’un certain seuil de lacunes et d’atrophie cérébrale.
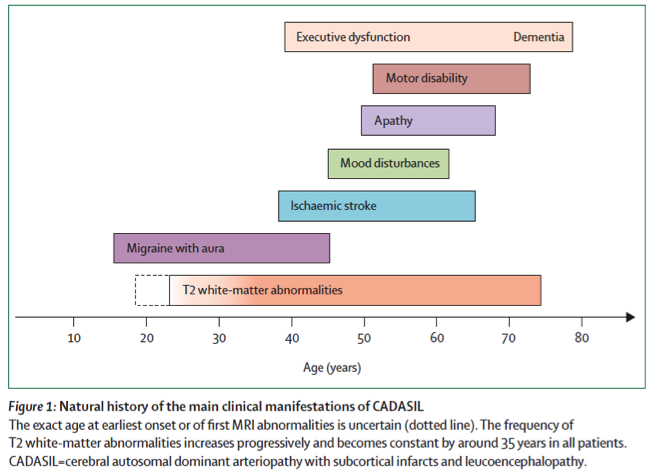
Figure 1: Histoire naturelle résumée de la maladie (source: Lancet Neurol 2009; 8: 643–53)
Diagnostic
L’imagerie par résonance magnétique (IRM) est essentielle au diagnostic de la maladie. Les anomalies de signal en IRM (anomalies de la substance blanche cérébrale) sont parfois détectées avant la survenue des premiers symptômes de la maladie. Ces anomalies apparaissent entre l’âge de 20 et 35 ans et peuvent donc être encore inconstantes dans cette tranche d’âge. Par contre, après l’âge de35 ans, tous les sujets porteurs du gène muté, avec ou sans symptôme, ont des anomalies IRM évocatrices de la maladie. Leur absence totale après l’âge de 35 ans doit donc faire remettre en question le diagnostic.
Plusieurs types d’anomalies peuvent être observées (Figure 2).
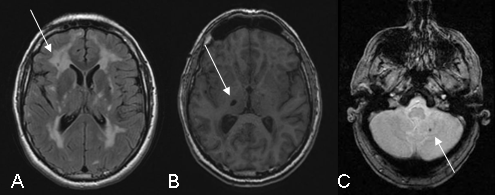
Figure 2: Illustration des anomalies IRM rencontrées sur les séquences FLAIR (A), T1 (B) et écho de gradient (C)
Les Hypersignaux de la substance blanche (A) sont constants en présence de symptômes de la maladie. Ils sont observés sur les séquences pondérées en T2 qui montrent de vastes étendues hyperintenses au sein de la substance blanche associées à des anomalies plus focales au sein des noyaux gris, du thalamus et du tronc cérébral. L’étendue des hypersignaux de la substance blanche est variable et augmente avec l’âge. Chez les sujets de moins de 40 ans, les anomalies de signal sont habituellement punctiformes ou nodulaires et de répartition symétrique. Progressivement au cours de l’évolution de la maladie, les hypersignaux deviennent confluents et s’étendent à toute la substance blanche. La présence de ces anomalies de signal au niveau des pôles antérieurs des lobes temporaux (plus de 2 patients sur 3) a une valeur diagnostique très importante en particulier lorsqu’elles sont associés à la présence d’espaces périvascualaires dilatés à la jonction cortico-souscorticale (Figure 3). Cette atteinte du pôle tout antérieur des lobes temporaux n’est pas retrouvée dans les maladies artériolaires cérébrales dues à l’hypertension artérielle ou au diabète.
Les infarctus lacunaires (B) sont détectés sur les images pondérées en T1 sous la forme de zones limitées en hyposignal. Ils sont punctiformes ou plus larges en rapport avec la cavité se formant secondairement après la survenue d’un petit infarctus. Ces lésions sont observées chez environ deux patients sur trois ayant des anomalies de la substance blanche.
Ils sont présents au sein de la substance blanche, des noyaux gris et du tronc cérébral. Le volume total de ces lésions est fortement corrélé à la sévérité clinique de la maladie.
Des microsaignements (C) sont mis en évidence chez un patient sur trois en moyenne à l’aide de séquences pondérées en écho de gradient ou T2* très sensibles à l’accumulation des dérivés de l’hémoglobine dans le tissu cérébral. Ces saignements ne sont habituellement pas responsables de symptomes spécifiques mais leur présence semble le plus souvent associée à une atteinte plus sévère de la paroi vasculaire et de la maladie. Ils peuvent être localisés dans les régions profondes ou juxtacorticales des hémisphères cérébraux, au niveau du tronc cérébral ou du cervelet.
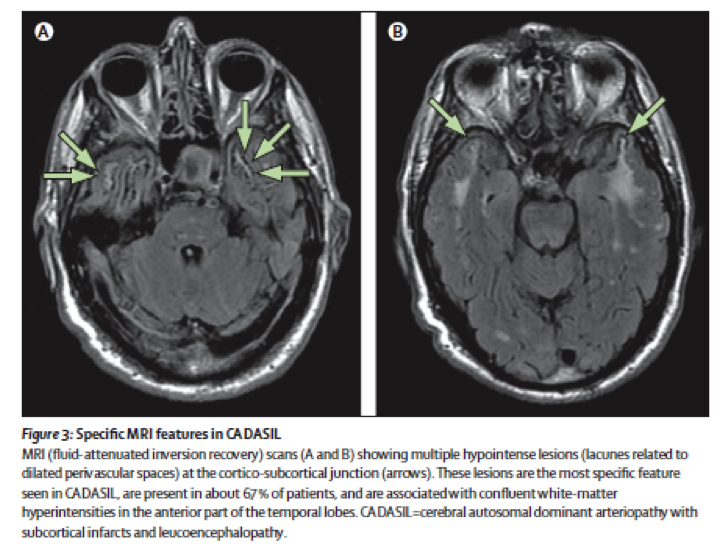
Figure 3: Anomalies IRM spécifiques de CADASIL
Diagnostic
CADASIL est une maladie familiale héréditaire. Le mode de transmission est autosomique dominant (atteinte de même fréquence des sujets de sexe masculin et féminin, 50% des enfants issus d’un sujet atteint ayant l’anomalie génétique) (Figure 4).
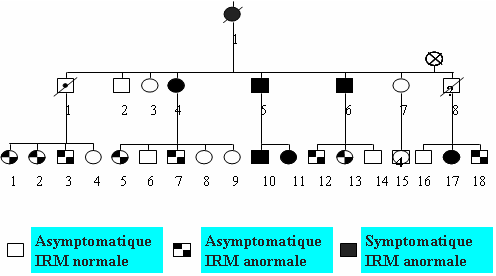
Figure 4: Arbre généalogique au sein d’une famille montrant la transmission de type autosomique dominant et les résultats de l’examen IRM.
Le diagnostic doit être évoqué chez les patients ayant des lésions symétriques de la substance blanche et une histoire clinique de crises migraine avec aura, d’accidents ischémiques cérébraux transitoires ou constitués, de troubles de l’humeur ou de troubles cognitifs d’origine inexpliqués.
L’interrogatoire à la recherche d’antécédents évocateurs chez d’autres membres de la famille est indispensable. Une histoire de sclérose en plaques (le diagnostic erroné de sclérose en plaques est parfois évoqué chez les sujets jeunes après un premier évènement clinique), d’accidents vasculaires cérébraux ou de démence progressive avec des troubles moteurs chez des apparentés doivent faire évoquer une maladie familiale des petits vaisseaux. Il faut noter cependant que l’absence totale d’antécédent familial ne permet pas d’écarter le diagnostic en raison de la possibilité de mutation « de novo » du gène responsable à l’origine de cas sporadique.
La présence d’anomalies de signal, sur les séquences T2 ou FLAIR en IRM, à distribution symétrique, au niveau de la substance blanche, en particulier au niveau de la partie antérieure des lobes temporaux augmente la probabilité diagnostique de CADASIL en raison de leur spécificité.
Le bilan à la recherche d’une autre cause d’atteinte des artères cérébrales de petit calibre : bilan biologique simple, recherche d’un syndrome inflammatoire, de facteurs de risque vasculaire (bilan lipidique, homocystéinémie, glycémie à jeun, holter tensionnel), écho-doppler des vaisseaux du cou et transcrânien, est habituellement négatif.
Lorsque le diagnostic est suspecté, l’artériographie cérébrale (par voie intraveineuse) doit être évitée car elle peut être à l’origine de manifestations neurologiques sévères (céphalées importantes, crises de migraine avec aura sévère) parfois graves. Cet examen est le plus souvent normal, il peut montrer parfois un rétrécissement des petites artères. Une angiographie par résonance magnétique doit être préférée pour visualiser l’état des artères de moyen ou de gros claibre.
 Pour confirmer le diagnostic, un test génétique sera réalisé dans tous les cas. Le gène impliqué est le gène Notch3, situé sur le bras court du chromosome 19. Il est constitué de 33 exons dont 23 exons (de 2 à 24) codent pour des motifs EFG-like qui comportent six résidus cystéine. A ce jour, toutes les mutations responsables de la maladie ont été localisées dans ces exons (exons 2 à 24). Ces mutations sont très stéréotypées et conduisent toutes au gain ou à la perte d’une cystéine dans un des motifs EGF-like. La présence d’une mutation de ce type permet d’affirmer le diagnostic de la maladie avec certitude. Dans la population française, dans 70% des cas la mutation siège dans les exons 3 ou 4 du gène Notch3 et dans 90 à 95% des cas la mutation siège dans l’un des 12 exons suivants 2, 3, 4, 5, 6, 7, 8, 11, 12, 18, 19 ou 20. En l’absence de mutation connue dans la famille du sujet atteint, les exons 3 et 4 (sensibilité 70%) sont d’abord testés, puis les exons 2, 5, 6, 7, 8, 11, 12, 18, 19 ou 20 (sensibilité 95%). Si les arguments en faveur du diagnostic sont très forts (importance de l’envoi des données cliniques et IRM) et si l’analyse précédente est négative, le criblage peut être étendu aux derniers exons du gène qui sont mutés chez un pourcentage très limité de patients CADASIL. La sensibilité estimée du criblage des 23 exons codant pour les domaines EGF de Notch 3 est proche de 100 %.
Pour confirmer le diagnostic, un test génétique sera réalisé dans tous les cas. Le gène impliqué est le gène Notch3, situé sur le bras court du chromosome 19. Il est constitué de 33 exons dont 23 exons (de 2 à 24) codent pour des motifs EFG-like qui comportent six résidus cystéine. A ce jour, toutes les mutations responsables de la maladie ont été localisées dans ces exons (exons 2 à 24). Ces mutations sont très stéréotypées et conduisent toutes au gain ou à la perte d’une cystéine dans un des motifs EGF-like. La présence d’une mutation de ce type permet d’affirmer le diagnostic de la maladie avec certitude. Dans la population française, dans 70% des cas la mutation siège dans les exons 3 ou 4 du gène Notch3 et dans 90 à 95% des cas la mutation siège dans l’un des 12 exons suivants 2, 3, 4, 5, 6, 7, 8, 11, 12, 18, 19 ou 20. En l’absence de mutation connue dans la famille du sujet atteint, les exons 3 et 4 (sensibilité 70%) sont d’abord testés, puis les exons 2, 5, 6, 7, 8, 11, 12, 18, 19 ou 20 (sensibilité 95%). Si les arguments en faveur du diagnostic sont très forts (importance de l’envoi des données cliniques et IRM) et si l’analyse précédente est négative, le criblage peut être étendu aux derniers exons du gène qui sont mutés chez un pourcentage très limité de patients CADASIL. La sensibilité estimée du criblage des 23 exons codant pour les domaines EGF de Notch 3 est proche de 100 %.
L’évolution des techniques de séquençage a maintenant supplanté l’étude de la biopsie de peau dans le diagnostic de la maladie. La biopsie cutanée n’est donc réalisée actuellement que lorsqu’il persiste un doute diagnostique à l’issue du séquençage des 23 exons du gène NOTCH3, ce qui est très rare ou en cas d’identification d’un variant de séquence n’impliquant pas un résidu cystéine et de signification inconnue. En cas de réalisation d’une biopsie de peau, un examen en microscopie électronique est réalisé à la recherche de matériel granulaire osmiophile (GOM) au niveau de la paroi vasculaire.
Le diagnostic génétique est possible avant l’apparition des symptômes de la maladie pour les membres d’une famille atteinte. Cependant, la réalisation d’un test génétique chez un sujet sain n’ayant aucune manifestation clinique de la maladie et n’ayant eu aucun examen préalable n’est effectuée que dans le cadre d’une consultation spécialisée et multidisciplinaire. Après une évaluation neurologique (neurologue), psychologique (entretien avec un ou une psychologue) et une consultation de génétique (généticien), la demande du patient est évaluée de façon collégiale et un délai de réflexion de plusieurs semaines est proposé avant la réalisation d’un prélèvement sanguin. Le choix de ne pas être informé des résultats du test est préservé tout au long de la procédure, jusqu’à l’annonce des résultats. Un suivi clinique et psychologique est toujours proposé après l’annonce des résultats.
Aucun test génétique n’est actuellement réalisé chez les sujets mineurs asymptomatiques.
Etiologie/physiopathologie
Les symptômes de la maladie sont principalement dus aux lésions survenant au niveau du cerveau au cours de la maladie. Les mécanismes à l’origine des hypersignaux de la substance blanche observées en IRM sur les séquences T2 et FLAIR et que l’on nomme leucoencéphalopathie sont imparfaitement connus. Une démyélinisation (perte des gaines de myéline fabriquées par les oligodendrocytes dans la substance blanche) et une perte des axones des neurones du cerveau en rapport avec une diminution de la perfusion cérébrale est le mécanismes habituellement évoqué. Mais d’autres mécanismes sont probablement impliqués. En effet, une étude récente a montré que l’étendue de ces hypersignaux apparait liée à une augmentation du volume cérébral et non à l’atrophie cérébrale. Ces résultats suggèrent que la leucoencéphalopathie observée dans CADASIL pourrait être liée en partie à une augmentation du contenu en eau de certaines régions du cerveau. Cette leucoencéphalopathie est associée à de petits infarctus situés surtout en profondeur du cerveau et dus à l’interruption du flux sanguin dans une zone irriguée par une petite artère. Les infarctus peuvent laisser comme séquelle une petite cavité ou trou appelés « lacune ». Des traces de toutes petites hémorragies peuvent aussi être visibles chez un tiers des patients. Les études en imagerie cérébrale montrent que c’est principalement, l’accumulation des petits infarctus au niveau cérébral qui explique la gravité de la maladie au cours de CADASIL.
CADASIL est une maladie qui touche principalement la paroi des petites artères (artérioles) du cerveau (mais aussi des autres organes). La paroi des artères est souvent épaissie, parfois fibreuse. Les cellules musculaires lisses au niveau de la couche centrale de la paroi du vaisseau (média) sont anormales et ou en voie de disparition. A leur voisinage, on peut observer un matériel granuleux appelé GOM (granular osmiophilic material), tout à fait caractéristique de la maladie et visible en microscopie électronique. Des travaux chez l’homme mais aussi chez les souris ayant l’anomalie génétique ont montré que la paroi des petites artères ne se contractait pas ou ne se dilatait pas normalement. Il est possible que le rétrécissement de certains vaisseaux et cette réactivité anormale de la paroi expliquent le défaut d’irrigation observée au cours de cette affection.
Dans CADASIL, les mutations du gène NOTCH3 aboutissent à une accumulation dans la paroi du vaisseau d’une partie de la protéine mutée (domaine extracellulaire du récepteur NOTCH3) et à une aggrégation à proximité de protéines de la matrice extracellulaire (qui constituent les GOM observés en microscopie électronique). La raison pour laquelle les cellules musculaires lisses fonctionneent anormalement et dégénèrent est encore inconnue. Le rôle important du gène Notch3 dans le développement des petites artères est par contre clairement démontré.
Prise en charge
Aucun traitement préventif spécifique de la maladie n’est à ce jour connu dans CADASIL. En raison de la survenue d’infarctus cérébraux, l’aspirine est classiquement utilisée en prévention secondaire mais le bénéfice de ce traitement au cours de la maladie n’a pas été démontré. La survenue possible d’hémorragies intracrâniennes, même si elles sont rares suggère que l’utilisation d’anticoagulants serait par contre risquée.
Pour les crises de migraine avec aura, les vasoconstricteurs sont déconseillés en raison du risque théorique de réduction du débit sanguin cérébral chez des patients présentant un état hémodynamique précaire et une baisse du débit sanguin cérébral. Les anti-inflammatoires non stéroïdiens et les antalgiques sont donc recommandés en première ligne pour traiter les crises de migraine.
L’intérêt des inhibiteurs de l’acétylcholinestérase a été évalué récemment pour améliorer les patients ayant des troubles cognitifs. Cette étude s’est avérée négative pour le critère de jugement principal. Certaines analyses complémentaires suggèrent un effet favorable du donezepil sur les troubles des fonctions exécutives observés au cours de CADASIL mais devront être confirmés par de nouvelles études.
Tous les traitements hypotenseurs (neuroleptiques, anti-hypertenseurs) doivent être employés avec prudence en raison du risque éventuel de baisse du débit sanguin cérébral chez des patients ayant une perfusion du cerveau réduite.
Par contre, la kinésithérapie et la rééducation sont indispensables et doivent être prescrits largement lorsque des séquelles motrices, des troubles de la marche et de l’équilibre sont apparus, en particulier après la survenue d’un accident vasculaire cérébral. L’orthophonie est prescrite pour améliorer les capacités de communication et de langage lorsque cela est nécessaire.
Le soutien psychologique est crucial à toutes les étapes de la maladie, pour le patient mais aussi pour la famille et les accompagnants. Il comprend à la fois la prise en charge des conséquences psychologiques liées au déficit neurologique, l’évaluation des troubles psychologiques directement dus à la maladie, la prise en charge des conséquences du handicap au niveau familial, le soutien psychologique en raison du caractère familial et héréditaire de la maladie.
Etat de la recherche
 Les travaux de recherche actuels s’orientent actuellement dans deux directions :
Les travaux de recherche actuels s’orientent actuellement dans deux directions :
1) sur le plan clinique, les études de suivi de patients ayant un CADASIL ont permis de préciser les paramètres cliniques et IRM qui expliquent la variabilité de la sévérité de la maladie. Ceci devrait permettre la mise en place d’un essai thérapeutique dans les années à venir. 2) l’étude des mécanismes moléculaires conduisant de l’anomalie génétique du gène NOTCH3 aux lésions observées au sein de la paroi du vaisseau qui se développe grâce à des modèles animaux de la maladie.